Workflow settings#
Workflow setting are in config.yml:
Configuration file
orfeome_name: uORFbarcodes
# Conditions in experiment
# Sample files should be in reads/ directory
# and have this format condition_1.fastq.gz
# _1 represents the bin number of that condition
conditions:
test: [Test]
control: [Control]
# Number of bins set during sorting of cells
# (should be the same for each sample)
# With bin_number higher than 1, it is assumed that the user wants to perform
# a protein stability analysis using PSI (Protein Stability Index) as a metric.
# With bin_number = 1, the user wants to perform a pairwise comparison
# of ORF counts between two conditions using MAGeCK/DrugZ.
bin_number: 6
cutadapt:
# 5' adapter sequence to trim
# Use "" to disable 5' adapter trimming
g: CCAGTAGGTCCACTATGAGT
# 3' adapter sequence to trim
# Use "" to disable 3' adapter trimming
a: AGCTGTGTAAGCGGAACTAG
# Trim <INT> nucleotides (before a/g trimming)
# If positive, remove bases from the beginning
# If negative, remove bases from the end.
u: 0
# Shorten reads to <INT> nucleotides
# Positive values remove bases at the end
# negative values remove bases at the beginning.
# This and the following modifications are applied after adapter trimming (a,b).
l: 20
# Extra arguments for cutadapt
extra: "-q 20 --discard-untrimmed"
csv:
# CSV file with the gene/ORF/barcode information
# 0-indexed column numbers (First column is 0)
gene_column: 4 # Column number with gene names
orf_column: 2 # Column number with unique ORF names
barcode_id_column: 0 # Column with unique barcode IDs
sequence_column: 1 # Column number with barcode sequences
# Alignment settings
bowtie2:
mismatch: 0 # mismatches allowed in the alignment
extra: "" # Extra arguments for bowtie2
# MAGeCK/DrugZ can be used when bin_number is set to 1
mageck:
run: True # Run MAGeCK analysis
extra_mageck_arguments: "--sort-criteria pos"
mageck_control_barcodes: all # All or file with control barcodes
fdr: 0.25 # FDR threshold for downstream mageck analysis
# DrugZ can be used when bin_number is set to 1
drugz:
run: True # Run DrugZ analysis
extra: "" # Extra DrugZ arguments
# Settings for the protein stability analysis when bin number is higher than 1
psi:
# Minimum value of sum of barcode counts across all bins to keep
sob_threshold: 100
# deltaPSI thresholds for hits
hit_threshold: [0.75, 1.0, 1.25]
# Exclude barcode with twin peaks
exclude_twin_peaks: True
# Proportion threshold for second peak of first peak
proportion_threshold: [0.5, 0.4, 0.35]
# Penalty factor for having less than median number of good barcodes
penalty_factor: [4, 4, 4]
# Barcode threshold for hits
# Keep ORFs with at least bc_threshold barcodes
bc_threshold: 2
# Multi condition heatmap settings
# Use R-style boolean values
heatmap:
# Show row names in the heatmap
rownames: TRUE
# Font size of the row names
row_font_size: 4
# Number of clusters for gene clustering
# This number is used for writing the clusters to a csv file
clusters: 6
# Height of the heatmap in inches
height: 8
# Width of the heatmap in inches
width: 4
Sample names#
The conditions section defines the conditions in the experiment. The sample files should be placed in the reads/ directory and should follow the naming convention <condition>_<bin_number>.fastq.gz, where <condition> is one of the conditions defined in the config.yml file (e.g. Test_1.fastq.gz, Control_1.fastq.gz, etc.).
conditions:
test: [Test]
control: [Control]
GPSW calculates \(dPSI\) values by pairing corresponding test and control conditions. For instance, if you define tests as [Test1, Test2] and controls as [Control1, Control2], GPSW will compare Test1 with Control1 and Test2 with Control2. It is essential that all samples share the same bin_number.
Bin number#
If the bin_number is set to 1, the workflow will perform a pairwise comparison of ORF counts between two conditions using MAGeCK/DrugZ. If the bin_number is greater than 1, the workflow will perform a protein stability analysis using Protein Stability Index (PSI) as a metric.
bin_number: 6
Cutadapt settings#
The cutadapt section defines the settings for trimming the raw reads. The options correspond to command line arguments for cutadapt. The g and a fields specify the 5’ and 3’ adapter sequences to trim, respectively. If you want to disable trimming, set these fields to an empty string (“”). The u field specifies the number of nucleotides to trim from the beginning or end of the reads, and the l field specifies the length to which the reads should be shortened. If l is positive, it removes bases from the end of the reads; if negative, it removes bases from the beginning.
Extra arguments for cutadapt can be specified in the extra field.
cutadapt:
# 5' adapter sequence to trim
# Use "" to disable 5' adapter trimming
g: CCAGTAGGTCCACTATGAGT
# 3' adapter sequence to trim
# Use "" to disable 3' adapter trimming
a: AGCTGTGTAAGCGGAACTAG
# Trim <INT> nucleotides (before a/g trimming)
# If positive, remove bases from the beginning
# If negative, remove bases from the end.
u: 0
# Shorten reads to <INT> nucleotides
# Positive values remove bases at the end
# negative values remove bases at the beginning.
# This and the following modifications are applied after adapter trimming (a,b).
l: 20
# Extra arguments for cutadapt
extra: "-q 20 --discard-untrimmed"
ORF library information#
Provide a CSV file with the ORF library information in resources/ directory. The CSV file should contain the following columns: ID, sequence, IOH_ID, and Gene_ID. See the example below:
ID |
sequence |
IOH_ID |
Gene_ID |
|---|---|---|---|
1_IOH10003_2802_PLD2 |
ATCCGAGTATAGAGACGTAAACTA |
IOH10003 |
PLD2 |
2_IOH10003_2802_PLD2 |
AACTACGTCATGAGCCGGATACCG |
IOH10003 |
PLD2 |
3_IOH10003_2802_PLD2 |
TTGCGCGCTGTGTTGTAACGTTAT |
IOH10003 |
PLD2 |
4_IOH10003_2802_PLD2 |
GACTAGGATGACTACGGAGTTTGC |
IOH10003 |
PLD2 |
5_IOH10003_2802_PLD2 |
GCGTCCTGTTATTCGTGATTGCGC |
IOH10003 |
PLD2 |
6_IOH10004_585_RAB22A |
ATACAGAGTAAGTTTCTCAAAATA |
IOH10004 |
RAB22A |
7_IOH10004_585_RAB22A |
CGGAGCATCTATTACAGAAAGGTA |
IOH10004 |
RAB22A |
In config/config.yml set the columns for this info as follows:
csv:
# CSV file with the gene/ORF/barcode information
# 0-indexed column numbers (First column is 0)
gene_column: 3 # Column number with gene names
orf_column: 2 # Column number with unique ORF names
barcode_id_column: 0 # Column with unique barcode IDs
sequence_column: 1 # Column number with barcode sequences
Alignment settings#
GPSW uses Bowtie2 for aligning the reads to the ORF library. The bowtie2 section defines the settings for the alignment. mismatch is the number of mismatches allowed in the alignment, and extra can be used to specify additional arguments for Bowtie2.
bowtie2:
mismatch: 0 # mismatches allowed in the alignment
extra: "" # Extra arguments for bowtie2
MAGeCK/DrugZ settings#
When bin_number is set to 1, the workflow runs MAGeCK/DrugZ. The mageck section defines the settings for the MAGeCK analysis. The run field specifies to run MAGeCK/DrugZ analysis, and extra_mageck_arguments can be used to specify additional arguments for MAGeCK. The mageck_control_barcodes field specifies whether to use all control barcodes or a file with control barcodes. The fdr field specifies the FDR threshold for downstream MAGeCK analysis.
mageck:
run: True # Run MAGeCK analysis
extra_mageck_arguments: "--sort-criteria pos"
mageck_control_barcodes: all # All or file with control barcodes
fdr: 0.25 # FDR threshold for downstream mageck analysis
drugz:
run: True # Run DrugZ analysis
extra: "" # Extra DrugZ arguments
PSI settings#
The variables that control the PSI analysis are defined in the psi section of the config.yml file. The PSI analysis is performed when bin_number is greater than 1, and it calculates the Protein Stability Index (PSI) for each ORF based on the proportion of reads across multiple bins.
The values between square brackets (e.g. [0.75, 1.0, 1.25]) indicate that the workflow will run the analysis for each value in the list, allowing for multiple thresholds to be applied in the analysis. The results will be saved in separate files for each threshold.
Note
All of the values in brackets are lists, and all of these must have the same length (i.e. the same number of values).
psi:
# Minimum value of sum of barcode counts across all bins to keep
sob_threshold: 100
# deltaPSI thresholds for hits
hit_threshold: [0.75, 1.0, 1.25]
# Exclude barcode with twin peaks
exclude_twin_peaks: True
# Proportion threshold for second peak of first peak
proportion_threshold: [0.5, 0.4, 0.35]
# Penalty factor for having less than median number of good barcodes
penalty_factor: [4, 4, 4]
# Barcode threshold for hits
# Keep ORFs with at least bc_threshold barcodes
bc_threshold: 2
# SD threshold for most stringent hits
# mean deltaPSI > sd_threshold * SD
sd_threshold: [2, 2, 2.5]
More on the PSI analysis can be found in the Background section.
bin_number#
When bin_number is set to 1, the workflow performs a pairwise comparison of ORF counts between the test and control conditions using MAGeCK and/or DrugZ (see Single-bin screen analysis (bin_number: 1)). The psi section settings are ignored in this mode.
When bin_number is greater than 1, the workflow performs a protein stability analysis using PSI as a metric. The psi section defines the settings for the PSI analysis.
sob_threshold#
The sob_threshold is the minimum value of the sum of barcode counts across all bins to keep an ORF (100 is recommended).
hit_threshold#
The hit_threshold defines the \(dPSI_i\) thresholds value for calling a hits. For example, if the hit_threshold is set to 0.75, then an ORF will be considered a hit if its \(dPSI_i\) value is greater than 0.75.
proportion_threshold#
The proportion_threshold is used in the twin peaks analysis. It defines the minimum proportion of the second peak relative to the first peak for a barcode to be considered a twin peak. For example, if the proportion_threshold is set to 0.5, then a barcode will be considered a twin peak if its second peak is at least 50% of the first peak.
Note
Good barcodes are defined as those which do not have a twin peak in the distribution of their counts across bins. Barcodes with twin peaks are defined as having two peaks that are at least two bins apart (\(\Delta Bin > 1\)) and the second peak has to be a minimum proportion of the highest peak. This proportion is defined by the user in the config.yml file (proportion_threshold). See the example below for a visual representation of this. Not all twin peaks are marked in this example.
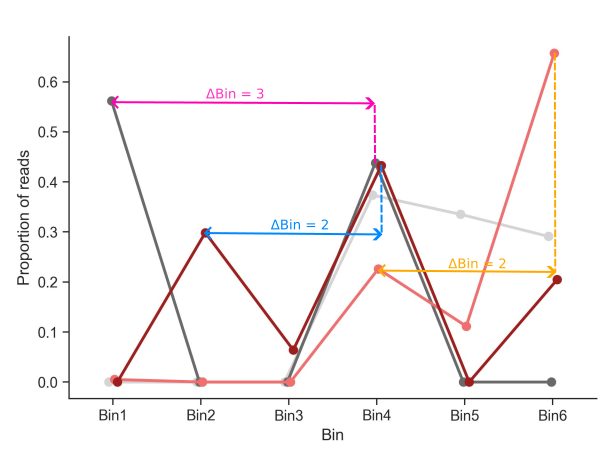
Twin peaks example#
penalty_factor#
The penalty_factor is a list of values that defines the penalty factor for having less than the median number of good barcodes. This is used to penalize ORFs with fewer good barcodes, which can affect the reliability of the PSI analysis.
The penalty_factor is applied as follows:
Where:
\(z_{c}\) is the corrected \(z\).
\(z\) is the z-score.
\(n\) is the number of good barcodes.
\(m\) is the median of good barcodes of all ORFs.
\(p\) is a user-defined penalty factor (
penalty_factorin config.yml).
This correction applies a mild penalty to the z-score of ORFs with fewer good barcodes, which helps to account for the reduced reliability of the PSI analysis in those cases. The recommended value for penalty_factor is 4, but it can be adjusted based on the specific requirements of the analysis (a lower value gives a higher penalty).
bc_threshold#
The bc_threshold is the minimum number of ‘good’ barcodes required to keep an ORF.
heatmap#
When multiple conditions are present, the workflow generates a heatmap of \(dPSI_i\) values for each ORF found as hits in either test conditions. The heatmap section defines the settings for the heatmap generation.
heatmap:
# Show row names in the heatmap
rownames: TRUE
# Font size of the row names
row_font_size: 4
# Number of clusters for gene clustering
# This number is used for writing the clusters to a csv file
clusters: 6
# Height of the heatmap in inches
height: 8
# Width of the heatmap in inches
width: 4
The clusters parameter defines the number of clusters for gene clustering. The best value for this depdends on how many comparisons are present and if all comparisons have proteins stabilised/destabilised. For example, with two comparisons that both have proteins stabilised and destabilised, a value of 6 is recommended:
Both comparisons have proteins stabilised.
Both comparisons have proteins destabilised.
One comparison has proteins stabilised, the other has proteins destabilised/no effect/not found.
One comparison has proteins stabilised, the other has proteins stabilised/no effect/not found.
One comparison has proteins destabilised, the other has proteins destabilised/no effect/not found.
One comparison has proteins destabilised, the other has proteins stabilised/no effect/not found.
If there are more comparisons or not all comparisons have proteins stabilised/destabilised, the number of clusters has to be adjusted accordingly.